在亨廷顿病(Huntington disease,HD)中,亨廷顿蛋白(HTT)N端聚谷氨酰胺结构域异常扩增(polyQ,>37Q),导致HTT发生错误折叠并聚集在神经元的突起和末端。细胞清除错误折叠蛋白主要有两条途径:泛素蛋白酶体系统和自噬溶酶体途径。其中,K48连接的泛素化主要负责引导蛋白质进入蛋白酶体进行降解,而K63连接的泛素化除介导蛋白质之间的相互作用外,也可参与自噬等多种胞内功能。课题组前期曾在老年猴脑中发现泛素结合酶UBE2N随衰老表达升高及蛋白酶体活性的受损,UBE2N可通过K63位泛素化修饰促进突变亨廷顿的积累(PNAS,2014;HUM MOL GENET,2015)。近期在阿尔茨海默病小鼠和患者脑中,发现淀粉样β蛋白生成期间UBE2N水平升高,抑制UBE2N可改善Aβ生成,其机制会涉及K48位泛素化的UPS途径清除(ALZHEIMERS DEMENT,2024)。2025年8月20日,暨南大学粤港澳中枢神经再生研究院李晓江和殷鹏团队在《自噬》杂志发表题为“Inhibition of UBE2N promotes the clearance of mutant HTT in HD knock-in mice”的研究论文。在该研究中,作者发现在亨廷顿病敲入(HD KI)小鼠脑中通过抑制UBE2N,可减少突变HTT蛋白的聚集,并鉴定了两种泛素特异性蛋白酶—USP29和USP49在此过程中的作用。

在本研究中,作者首先利用蛋白酶体和自噬抑制剂抑制细胞降解功能,发现会导致可溶性和聚集性的突变HTT增加,抑制自噬会使HTT聚集体进一步增多。在HD KI小鼠纹状体中,作者证实蛋白酶体系统的谷氨酰胺后和胰凝乳蛋白酶样的活性保持稳定,这表明脑中突变HTT的清除存在缺陷。接下来,作者合成了针对小鼠UBE2N基因的反义寡核苷酸ASOs及特异性抑制剂NSC697923,对HD KI小鼠进行为期四周的干预。分析显示抑制UBE2N可减少突变HTT蛋白的聚集,但不影响野生型HTT水平,同时与HTT聚集相关的K63泛素化修饰减少。为探究阻断UBE2N对HTT聚集物的清除机制,作者首先评估了自噬标记物,但BECN1、LC3和SQSTM1/p62并无显著变化;而K48介导的泛素化水平以及谷氨酰胺后和胰凝乳蛋白酶样活性均有所升高,这表明通过对多泛素链类型的改变和蛋白酶体活性的提升可能有助于促进突变HTT的降解。因此,作者对经NSC697923处理的HD KI小鼠的纹状体组织进行了测序。结果显示,HD KI小鼠中上调的差异表达基因,尤其是泛素特异性蛋白酶(USPs)家族成员Usp29和Usp49可被NSC697923有效抑制。最后作者在HEK293和HeLa细胞中,敲低内源性USP29和USP49后K48位泛素化水平增强;同样在共同转染的120Q-HTT细胞系中,呈现出HTT聚集物的积累降低,但未影响23Q-HTT的表达水平。
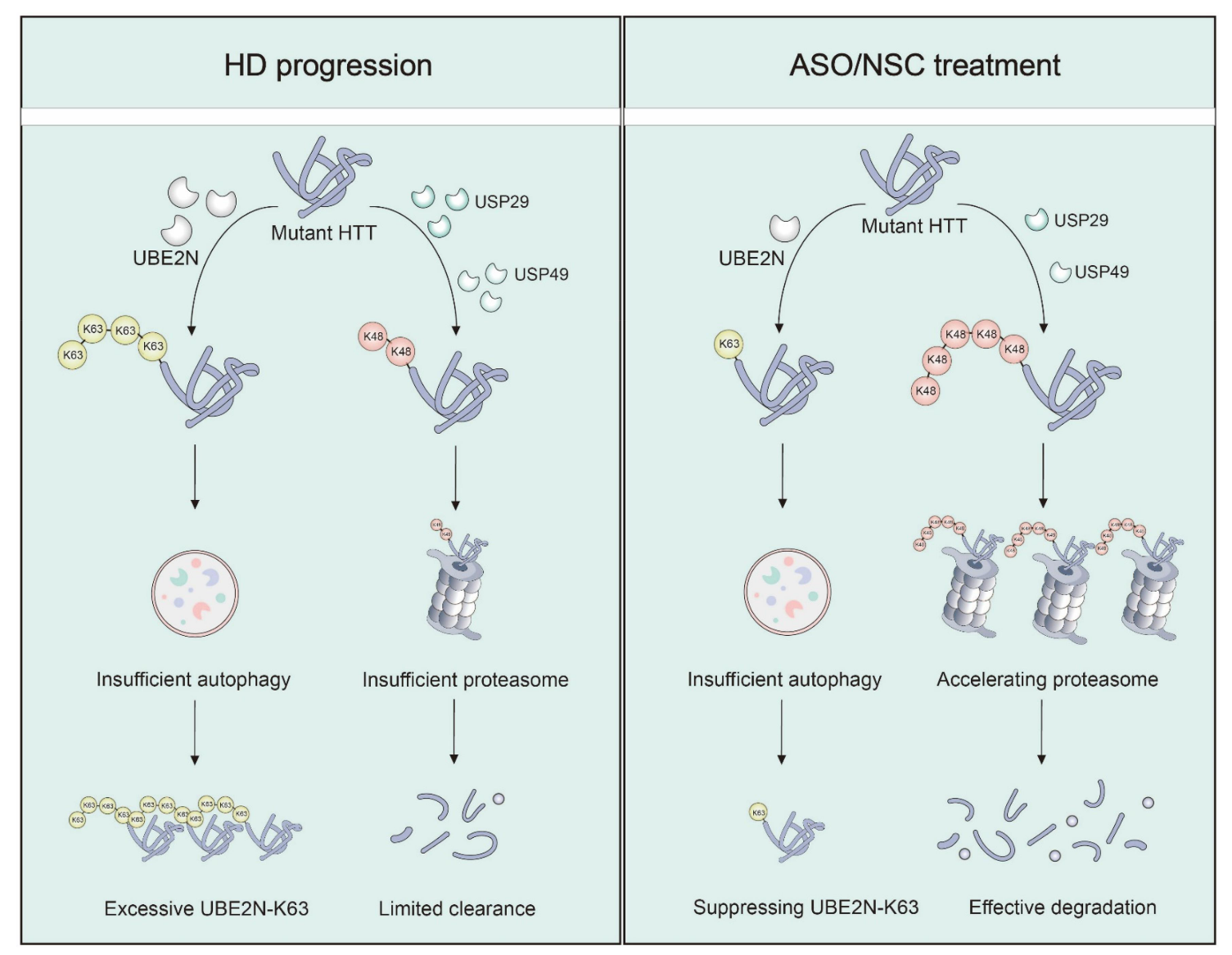
图:在HD病中,突变HTT与K48和K63多泛素链相连并积累。采用靶向ASOs或抑制剂NSC697923抑制UBE2N,可通过减少K63泛素化与Usp29、Usp49基因;增加K48泛素化与蛋白酶体活性而减少HTT聚集。
暨南大学的研究生欧凯丽和王翔为该论文的共同第一作者。李晓江教授和殷鹏研究员为该论文的共同通讯作者。本研究获得科技部重点研发计划,国家自然科学基金及广东省非人灵长类动物模型研究重点实验室平台的资助。
原文链接:https://doi.org/10.1080/15548627.2025.2549109