亨廷顿舞蹈症(Huntington’s Disease,HD)是一种常染色体显性遗传的神经退行性疾病,由位于4号染色体短臂上的Huntingtin (HTT)基因中CAG三核苷酸重复序列异常扩增引起,临床为运动障碍、认知功能下降和精神行为异常。突变型亨廷顿蛋白(mutant Huntingtin,mHTT)携带过长的多聚谷氨酰胺(polyQ)链,赋予mHTT毒性功能,但其如何导致纹状体中型多棘神经元(medium spiny neurons, MSNs)的选择性死亡,至今尚未完全阐明。
大量证据已揭示HD伴随着全局性的转录稳态失衡及DNA甲基化修饰异常。然而,这些表观遗传改变是疾病的驱动因素还是伴随现象,其深层分子通路亦有待阐明。此外,传统小鼠模型在模拟人类HD典型神经退行性病变方面的局限性,更增添了研究复杂性。上述科学问题长期制约着该领域的理论突破与临床转化。
2026年6月19日,暨南大学粤港澳中枢神经再生研究院林莉、李世华和李晓江团队在《Cell Reports》上发表了题为“Mutant Huntingtin disrupts TET1 transcription and alters DNA methylation in a Huntington‘s disease knock-in pig model”的研究论文 (https://www.cell.com/cell-reports/fulltext/S2211-1247(26)00662-5)。

该研究基于团队此前建立的HD基因敲入(knock-in, KI)猪模型(Cell, 2018),首次揭示mHTT通过异常结合转录因子TBP,抑制DNA去甲基化酶TET1的转录,导致全基因组5-甲基胞嘧啶(5mC)与5-羟甲基胞嘧啶(5hmC)稳态失衡的分子机制。
研究团队发现,HD-KI猪脑组织中5mC水平显著升高,5hmC水平显著下降,纹状体变化最为明显,且具有脑区特异性的DNA甲基化重塑。差异甲基化基因富集于ATP结合、神经营养信号及长时程增强等神经元功能通路。在皮层、小脑和纹状体中,仅TET1表达显著降低,其余甲基化相关酶无明显变化。TET1下调在2月龄即可检测到,早于行为症状出现,提示其参与早期病理进程(图1)。而在HD小鼠中未观察到Tet1下调,表明该变化具有物种特异性。HD-KI猪模型中纹状体TET1和多脊神经元标志物DARPP32共染神经元明显减少,提示其可能与MSNs选择性易损性相关。
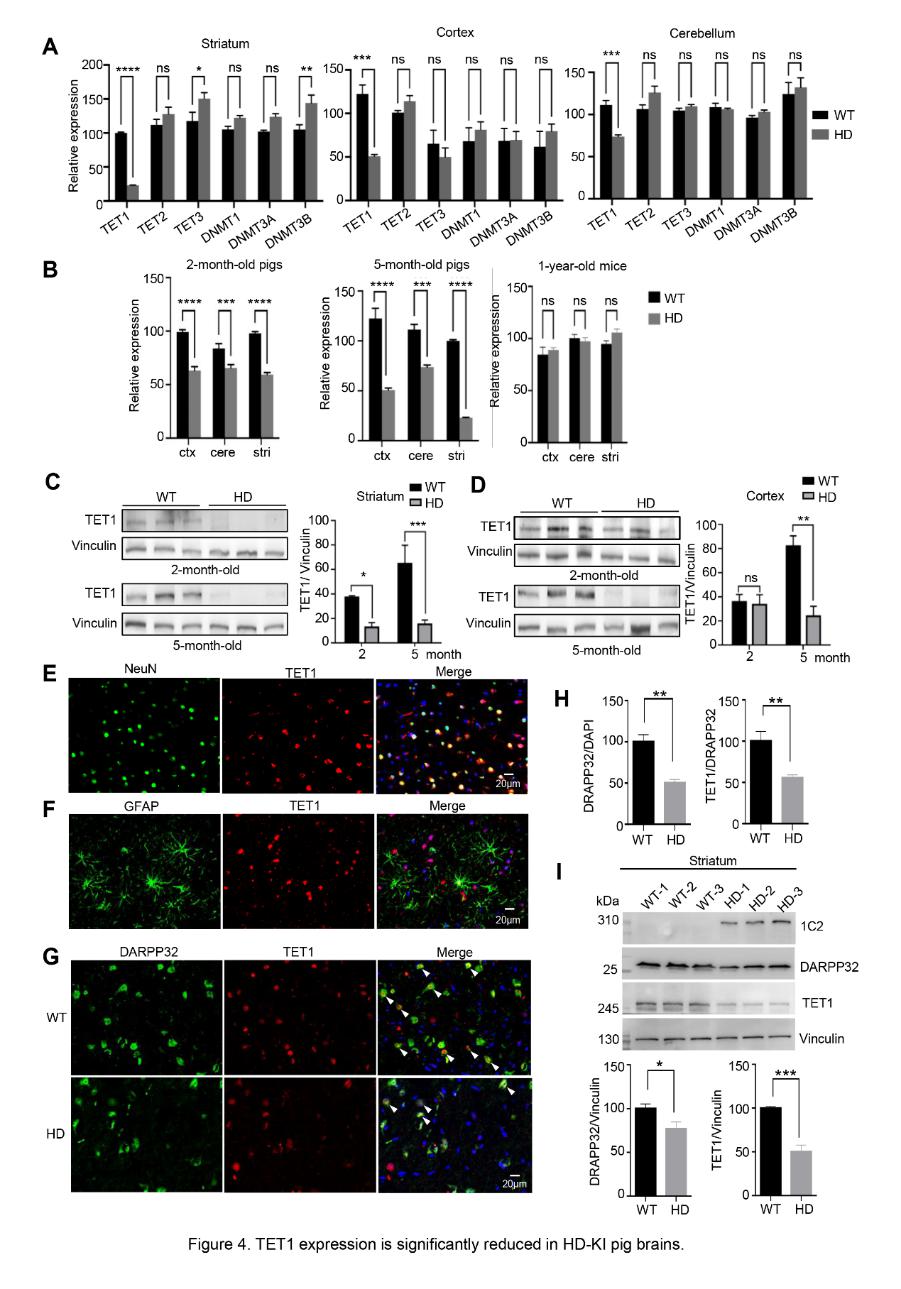
图1. TET1在HD-KI猪大脑中显著降低
上述致病通路在HD小鼠模型中并未呈现,其深层原因在于物种间启动子结构的差异性:人与猪的TET1启动子区域均富含TATA box核心调控元件,而小鼠TET1启动子则缺乏该关键序列。启动子序列比对分析显示,人源与猪源TET1启动子区域均富含TATA box核心基序,而小鼠Tet1启动子中几乎缺乏该调控元件。mHTT可显著抑制TBP介导的猪TET1启动子转录激活,但对小鼠Tet1启动子无显著影响,从分子层面解释了TET1下调的物种特异性。免疫共沉淀及组织学分析显示,HD猪脑组织中mHTT与TBP的相互作用显著增强,且TBP与mHTT聚集物存在明显的亚细胞共定位(图2)。
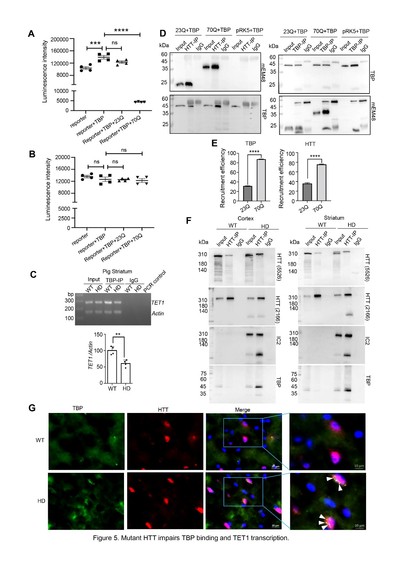
图2.突变HTT蛋白干扰转录因子TBP与TET1的结合,降低TET1表达。
本研究基于课题组自主构建的HD-KI猪模型,系统揭示了突变型亨廷顿蛋白(mHTT)通过异常结合通用转录因子TBP,进而抑制DNA去甲基化酶TET1转录的全新分子机制。该致病通路在HD小鼠模型中未能呈现,其根本原因在于物种间启动子结构的差异性:人源与猪源TET1启动子区域均富含TATA box核心调控元件,而小鼠Tet1启动子则缺乏该关键序列。
暨南大学粤港澳中枢神经再生研究院的博士研究生谢磊杰和刘小冬为文章的共同第一作者,林莉副研究员,李世华教授、李晓江教授和闫森研究员为共同通讯作者。本研究获得国家自然科学基金、广东省自然科学基金、生物活性分子与成药性国家重点实验室、广东省非人灵长类重点实验室等多项科研项目资助。